W tym miejscu należy zaznaczyć, że w przypadku molekuł rzeczywistych problem związku pomiędzy oddziaływaniami międzymolekularnymi a cechami makroskopowymi układu jest znacznie bardziej skomplikowany. Rzeczywiste molekuły zazwyczaj nie są symetryczne (w przeciwieństwie do cieczy prostych) a co za tym idzie orientacja momentu dipolowego może nie być bez znaczenia. W ramach prowadzonych badań przeprowadziliśmy szereg eksperymentów komputerowych mających na celu zbadanie wpływu wielkości jak i orientacji momentu dipolowego na stabilność termodynamiczną substancji przechłodzonych. W tym celu zaproponowano modelowy układy złożone z cząsteczek romboidalnych (RM) tzn. składających się z 4 atomów ułożonych w kształt rombu co sprawia, że odpowiednie rozłożenie ładunków elektrycznych na atomach pozwala wygenerować stały moment dipolowy o różnej orientacji – zorientowany wzdłuż dłuższej lub krótszej przekątnej rombu, co schematycznie zobrazowano na poniższym rysunku.
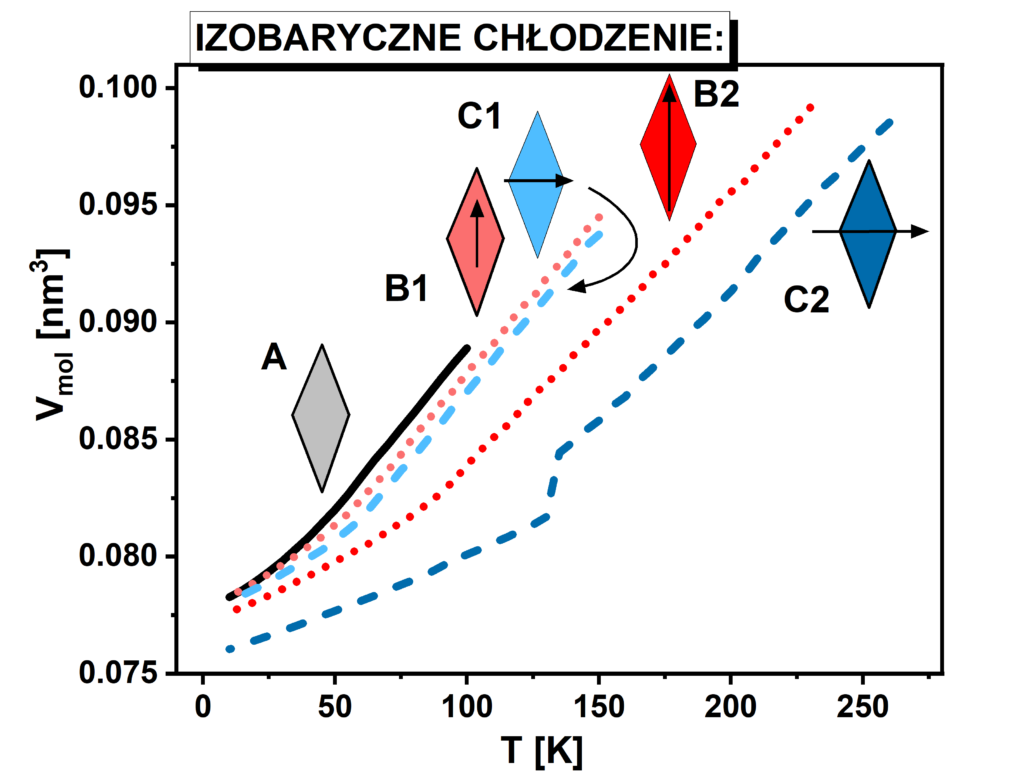
Na potrzeby eksperymentu przeprowadzono symulacje komputerowe odwzorowujące izobaryczne chłodzenie dla 5 modelowych układów. System A stworzony jest z RM bez momentu dipolowego, systemy B1 i B2 z RM posiadających mniejszy (1.61D) i większy (3.22D) moment dipolowy skierowany wzdłuż dłuższej przekątnej molekuły, podczas gdy systemy C1 i C2 analogiczne momenty dipolowe jednak zorientowane wzdłuż krótszej przekątnej molekuły. Jak widać na rysunku 1 przeprowadzone symulacje izobarycznego chłodzenia wykazały, że z pośród wszystkich badanych systemów tylko i wyłącznie jeden wykazuje skokową zmianę objętości, która świadczy o krystalizacji. Formowanie fazy krystalicznej w temperaturze 130K przez cząsteczki układu C2 potwierdzono poprzez analizę funkcji rozkładu radialnego jak również globalnego parametru porządku. Te ostanie są zaprezentowane na rysunku 2 gdzie dla układu C2 można zaobserwować ewidentny skokowy wzrost wartości globalnego parametru porządku.
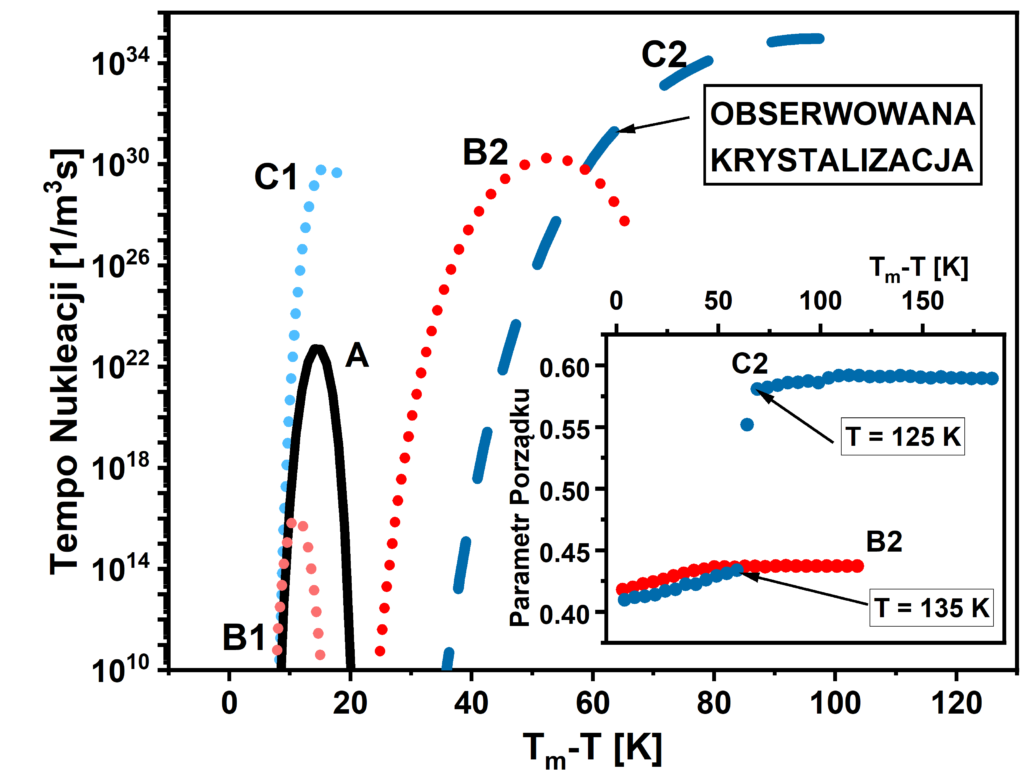
Zupełnie inne zachowanie przejawia układ B2. W tym przypadku wzrost wartości parametru porządku wynikający z ochładzania cieczy zostaje zatrzymany w pewnej temperaturze co świadczy o znacznym zahamowaniu dynamiki molekularnej i koresponduje z przejściem ciecz-szkło. Warto również zwrócić uwagę na fakt, że otrzymane wyniki mogą zostać wyjaśnione przy użyciu Klasycznej Teorii Nukleacji. Zgodnie z ogólnie akceptowanym opisem procesu krystalizacji rozpoczyna się on od powstania (w obrębie cieczy) zarodka kryształu (nukleacji), który następnie rośnie poprzez dołączanie do siebie kolejnych molekuł. Obie składowe procesu krystalizacji w racjonalny sposób opisują odpowiednio Klasyczna Teoria Nukleacji oraz model Normalnego Wzrostu Kryształu. Stworzenie zarodka wiąże się ze zmianą wartości energii swobodnej Gibbsa układu, ponieważ powstały kryształ charakteryzuje się mniejszą energią swobodną niż ciecz, z której został uformowany. Fakt ten sprzyja przemianie fazowej (jest to tzw. siła napędzająca krystalizację z ang. driving-force). Jednakże formowanie zarodka wymaga również wykonania pracy potrzebnej na stworzenie płaszczyzny oddzielającej kryształ od cieczy (interfejsu ciecz-kryształ). W konsekwencji zmiana energii swobodnej Gibbsa układu jest termodynamicznie kontrolowana przez współzawodnictwo pomiędzy siłą napędzającą krystalizację a wartością energii swobodnej interfejsu ciecz-kryształ a utworzenie stabilnego i trwałego zarodka mogącego zapoczątkować proces krystalizacji zarodka wiąże się z pokonaniem bariery energetycznej o konkretnej wartości. Nie jest to jednak jedyne ograniczenie, ponieważ przeorganizowanie molekuł prowadzące do powstania zarodka możliwe jest wyłącznie w sytuacji, gdy cząsteczki wykazują wystarczającą mobilność. Zatem w procesie zarodkowania kluczową rolę odgrywa również dyfuzja – przy dużych przechłodzeniach dynamika cząsteczek drastycznie maleje, co sprawia, że formowanie zarodków kryształów wewnątrz cieczy jest znacznie utrudnione. W konsekwencji stabilność termodynamiczna substancji zależy w pierwszej kolejności od tempa nuklecji czyli ilość stabilnych zarodków krystalicznych które mogą zostać stworzone w danej objętości substancji w konkretnym czasie. Jak można zaobserwować układ C2 charakteryzuje się największymi wartościami tempa nukleacji. Istotne jest również, że wartości tempa nukleacji, które osiąga układ C2 w temperaturze, w której krystalizuje nie są nigdy osiągane przez pozostałe modelowe układy. Zatem brak oznak krystalizacji pozostałych układów spowodowany jest niemożliwością uformowania się w nich zarodków krystalizacji.